NevGen probability Calculator of Time to Most Recent Common Ancestor
NevGen probability Calculator of Time to Most Recent Common Ancestor (further, just “TMRCA Calculator”) has been written in February 2013 (it’s core for calculation), but was available to several people who worked with it from December 2014, when user interface for it was written. Before that time, it worked only by entering haplotype data into source code.
Something must be made clear here, neither this nor any other program can not exactly calculate neither Time to Most Recent Common Ancestor nor number of generations to it. Because it is logicaly impossible, just like is impossible to calculate exactly how many passengers will enter any train in week from now. But in both cases is possible to calculate certain probabilities on basis of available statistical data. In case of train, on basis of several years’s statistics could be calculated probability that it has for example 235 passengers, or any other number.
This TMRCA Calculator uses different mutation probabilities for every STR marker. It uses Marko Heinila’s mutation rates, altough NevGen since 2015 has it’s own estimation of STR mutation rates (too lazy and time-short to implement it as another option in Calculator 🙂 ). Also, calculation takes into account backward mutations, and also multiple-step mutations. Mutation model gives about 5% of probability to two-step mutations (somewhere I found that frequency of two-step mutations is about 5%, I suppose in father-son pairs) and also some (much, much smaller) probabilities for 3- to 5-step mutation. Ofcourse, actual implemented mutation rates are normalised with this multistep mutation probabilities.
In left upper corner is edit field when descendant haplotypes should be entered, at most 500 of them, each in separate row. They may be in any of supported formats (FTDNA, 17 markers, 23 markers, see Description of NevGen Predictor tool), which is defined in list box in right side of Dialog Box.
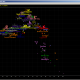
Now, let see small example. Suppose we want to see distribution of probabilities of Time to Most Recent Common Ancestor for two identical 12 marker haplotypes. We enter any 12 marker haplotype twice into input, and presuming MRCA had the same haplotype as both of descendants, also into “Haplotype of Ancestor” Field, and after calculation is done (it is slow, sorry :), I am too lazy to cache intermediate data to speed it up) we get this (Sample Image 01):

Output of calculation which is displayed is also automaticaly saved in file “TMRCA_results.txt”, in folder where executable is positioned, in Unicode TXT format.
Here in “Results” Field we can see calculated probabilities of TMRCA (for mutation model used in this Calculator) by number of generations, together with aggregate sum of it. In this sample there is 50% probability for TMRCA to be up to 16 generations. We do not see in this picture due to scrolling, but 90% of probability of TMRCA is at 54 generations. But, what if we put some STR marker value for MRCA different than for his two descendants? For example, we put for Ancestor (MRCA) here value of 14 for DYS393 (instead of 13). We got this (Sample Image 02):

Now in generated picture we can see quite different distribution. 50% probability from 16 now goes to 64 generations. Such case is, as we see, much less probable. Let suppose another scenario, we ofcourse do not know for sure exact value of DYS393 for Ancestor, and we want to calculate it without need to presume MRCA’s haplotype. In such case we need to check Field “Allowed unknown ancestor values” and make sure Field “Haplotype of Ancestor” is empty by erasing it’s contents. Then we got this (Sample Image 03):

There is not big difference here from Sample Image 01, but in some cases it might be. We can see that program automatically filled Field “Haplotype of Ancestor” with bunch of ‘$’, which mean that all STR values are not known. If we want, we might fix some of MRCA’s marker values, and other leave as unknown, like this (Sample Image 04):

Turning ON option “Allowed unknown ancestor values” is somewhat “dark side of Calculator” and is generally not recommended. Sometimes it can distort results, especially when there are more than two descendant haplotypes in calculation. Better is to somehow deduce MRCA’s haplotype and use it in calculation. If it is not entered by user, program shall automatically set it at average values of all entered descendant haplotypes. Please be aware that such “average values of all entered descendant haplotypes” need not be equal to actual haplotype of MRCA (Modal haplotype of entered descendant haplotypes). For that there is option “Determination of Ancestor’s haplotype by Treemaker”, which tries to deduce the most probable haplotype of MRCA (Modal haplotype), but for now it is not available, till its description is finished.
Next comes sample with two full 111-marker haplotypes (Sample Image 05):

There are 12 differences among them, and we got 50% of probability for 23 generations to the MRCA (ofcourse, it depends of which STR markers are different and how many, because they have different mutation rates). Setting all ancestor’s values to unknown does not change results much, we got then 50% of probability for 24 generations. Here we can see that distribution of probabilities is not so wide like it is with 12 markers. It is much narrower, as expected for increased precision with more markers. 90% of probability we got at 37 generations.
Exclusion of multicopy STRs
In some cases it could be very useful to exclude some or all multicopy markers from calculation. There are two cases for it.
The first case is general RecLOH mutation, which overwrites one STR marker copy over another. RecLOH mutation might involve several multicopy markers at the same time, which is contrary to Calculators’s presumtion of independence of mutations of different STRs. Even when RecLOH involves only one STR, it might make large steps, like mutation of 12-19 => 19-19. When RecLOH in any of descendant haplotypes is suspected, it is recommended to exclude it from calculation (however it might be safe and enough to exclude it only from haplotype where it is found, from others might not be needed). If such markers are not excluded, TMRCA would be exagerated.
-The second case is huge multistep mutation, when in descendant haplotypes exist very large differences between values of the same multicopy markers. Excelent example for this is haplogroup I2a1a M26, where at marker YCAII we have pairs 11-21, but also 21-21 or 11-11. Here we had huge multistep mutation from 11 to 21 (or vice versa), and trying to calculate probabilities of TMRCA of two haplotypes where one has 11-21 and another 21-21 will exagarate it. In such cases this marker should be excluded from calculation. RecLOH is also at work here, but maybe some other forces too.
There are two ways to solve this and exclude unwanted markers from calculation. The first is to put $ sign instead of unneeded STR values. This way, from next input:
13 24 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 1
13 24 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 2
13 24 14 11 14-14 11 12 12 13 13 29 19-19 ; Descendant 3, RecLOH on multiple markers is suspected to be at work here.
we got:
13 24 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 1
13 24 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 2
13 24 14 11 $ 11 12 12 13 13 29 $ ; Descendant 3, RecLOH on multiple markers is suspected to be at work here, involved markers are excluded from calculation.
Another way to solve this is to select “Single-value markers only”, in list box in right upper corner of Dialog Box. This way, all multimarker STRs shall be excluded from calculation, even those which are not problematic. This option also might be useful for testing purposes.
In the same list box there is another option “Multiple-value markers only”. It means that only multiple-value markers are used in calculation (single-value markers are ignored), and this can only be useful for testing purposes, when TMRCA is already known (see section “Results of testing of TMRCA Calculator”).
Ofcourse, default is option “Single and multiple-value markers” which means that all given markers are used in calculation, regardless of their number of copies.
TMRCA Calculator cannot work with haplotypes having different number of copies of the same STR, because probability of deletion or insertion of copies of STRs are not known to authors. For example, with such input data could not be calculated and aproppriate error message shall be displayed.
13 24 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 1
13 24-26 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 2
But, TMRCA Calculator can work with haplotypes having more than one copy of the same STR when there is usually one. For example, such input is regular, with multiple copies of DYS390:
13 24-25 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 1
13 24-26 14 11 11-14 11 12 12 13 13 29 15-19 ; Descendant 2
Other options in TMRCA Calculator
Option “Minimal number of markers” (right upper corner of Dialog Box) can be useful to users who handle great quantity of haplotypes (like us 🙂 ), and want to exclude from calculation haplotypes which are shorter from some length. Let suppose we have hundreds of haplotypes in TXT file, and we copy them all into Calculator. Some of them might be short (for example 12 markers) and we want to exclude them from calculation, but it is time consuming and error-prone to go through input data and erase them all. It is much more convenient to put, for example, 37 into this Field, and from calculation all short haplotypes shall be excluded, without need to erase them manually. Value of 0 means that all given haplotypes are used, regardless of length. In next sample we have three haplotypes (two with 37 a one with 12 markers, but this way we exclude short one from calculation), Sample Image 06:

In “Results” Field we can see that only two haplotypes are used, both with 37 markers.
In right side of Dialog Box there is list box which serves for user to select format and order of input haplotype data (“FTDNA order”, “17 markers”, in three different orders, and “23 markers”, in two different orders). But there are also options like “FTDNA order, only first 12 markers” (or 25, 37 or 67). Their meaning is somehow oposite to meaning of option “Minimal number of markers”. If we choose, for example, option “FTDNA order, only first 37 markers”, it’s effect is to exclude from calculation all markers above first 37. This is very useful for testing, and this can save a lot of time when preparing data when we have haplotypes with more of 37 markers but we want to test Calculator on exactly 37 markers, not more!
Option “Number of generations without common ancestor” is useful when user knows (or supposes) that input haplotypes has no common ancestor in certain number of generations. In that case, probabilities for given number of generations are set to zero and results are refreshed and displayed picture is redrawn, without slow recalculation of probabilities. Sample Image 07:

This is the same sample as Sample Image 05, except “Number of generations without common ancestor” is set to 18 instead of default 0. We can see now that aggregate 50% probability now goes to 26 generations.
IMPORTANT: NevGen TMRCA Calculator is not configured nor intended to calculate TMRCA probabilities too far away in past. It takes into consideration number of generations only up to 500. So, do not put haplotypes of different major haplogroups like R1a, R1b, I1, I2, J1, J2, E1b1b, G, N or O together into calculator. In such cases it shall not work properly nor give expected results. It is intended only to more recent number of generations.
Testing of TMRCA Calculator
TMRCA Calculator has been tested using some kind of “reverse technology”, using randomly generated artificial descendant haplotypes.
Full 111-marker haplotype was allowed to randomly mutate certain number of generations, using exactly the same mutation model as TMRCA Calculator is using. 600 of such independent descendant haplotypes were generated for several generation depths (20, 30, 40, 70, 80, 100, 150, 200 and 300 generations). This way were generated several sets of independent artificial descendants with known number of generations to common ancestor, whose haplotype is also known. Since artificial descendants were made using exactly the same mutation model as TMRCA Calculator is using, they are perfect to test it, since we know both haplotype of their MRCA and number of generations to it. And testing is done. Here shall be displayed some samples of it, using greater number of generated artificial descendants.
Let see the first such sample. We shall put first 100 haplotypes of 20 generations deep descendants (from file “ArtifDescendants\600h – 020 gener R1b 111 mrk.txt”, available in Tools), and in calculation we shall use all 111 markers. Sample Image 08:

We can see that 50% of probability goes to first 19.91 generations. And here is distribution very concentrated, which is also visible on picture. Next six numbers of generations were given almost all of probability.
Generations=18 probab = 1.10025% sum probab = 1.12073%
Generations=19 probab = 12.953% sum probab = 14.0737%
Generations=20 probab = 39.2076% sum probab = 53.2814%
Generations=21 probab = 35.0686% sum probab = 88.3499%
Generations=22 probab = 10.4477% sum probab = 98.7976%
Generations=23 probab = 1.15015% sum probab = 99.9478%
By checking Field “Allowed unknown ancestor values” we got very similar results and probability distribution, 50% goes to the first 19.7672 generations.
Another sample, this time with first 100 haplotypes of 70 generations deep descendants (from file “ArtifDescendants\600h – 070 gener R1b 111 mrk.txt”, available in Tools), and in calculation again all 111 markers, with checked Field “Allowed unknown ancestor values”. Sample Image 09:

The next picture calculates the same sample, but limited to only first 37 markers. Sample Image 10:

Here we got somewhat wider distribution of probabilities, due to less markers available to calculation.
Now we shall show several samples with first 200 haplotypes of 300 generations deep descendants (from file “ArtifDescendants\600h – 300 gener I2a1a 111 mrk.txt”, available in Tools). First calculation uses all 111 markers. Sample Image 11:

In the next picture is the same sample, but calculated using only multiple markers (16 of them in total). Sample Image 12:

Here we got 50% of probability to first 293.71 generations. Distribution of probabilities is wider, due to less markers used in calculation.
And next sample is the craziest, calculation using only foursome DYS464 (this is not available in user interface, and is not visible in report, I hade to modify source code manualy for this, to exclude from calculation all markers except DYS464). Sample Image 13:

Here we got 50% of probability to first 275.24 generations. Distribution of probabilities is much wider, due to less markers used in calculation (4×200 = 800). Do not mind for report, there says total of values (markers) is 3200, but it is 800, due to manual change of source code for this sample.
Final picture, using only first 12 markers. Sample Image 14:

Samples displayed here were not cherry-picked. To prove it, all sample sets were given in Folder “ArtifDescendants\”, so anyone can check himself any calculation he wants.
From many samples it can be concluded that TMRCA Calculator is biased to slightly increase number of generations with 50% of probability. This increase is less visible when number of generations till common ancestor is small (for example 20, or 70). It is more visible on artificially generated samples with depth of 300 generations. Bias is probably caused by accumulation of numerical error during calculation. Using by 500 artificially generated descendant haplotypes with all 111 markers and depth of up to 100 generations we found bias goes up to two generations, as can be seen from next table:
“600h – 020 gener I2a1a 111 mrk.txt” – 20.11 (20)
“600h – 020 gener R1b 111 mrk.txt” – 20.10 (20)
“600h – 030 gener I2a1a 111 mrk.txt” – 30.53 (30)
“600h – 030 gener R1b 111 mrk.txt” – 30.49 (30)
“600h – 040 gener I2a1a 111 mrk.txt” – 40.66 (40)
“600h – 040 gener R1b 111 mrk.txt” – 40.61 (40)
“600h – 070 gener I2a1a 111 mrk.txt” – 71.43 (70)
“600h – 070 gener R1b 111 mrk.txt” – 71.23 (70)
“600h – 080 gener I2a1a 111 mrk.txt” – 81.44 (80)
“600h – 080 gener R1b 111 mrk.txt” – 81.08 (80)
“600h – 100 gener I2a1a 111 mrk.txt” – 101.99 (100)
“600h – 100 gener R1b 111 mrk.txt” – 101.63 (100)
‘600h – 150 gener I2a1a 111 mrk.txt” – 155.18 (150)
“600h – 150 gener R1b 111 mrk.txt” – 154.46 (150)
“600h – 200 gener I2a1a 111 mrk.txt” – 207.34 (200)
“600h – 200 gener R1b 111 mrk.txt” – 206.37 (200)
“600h – 300 gener I2a1a 111 mrk.txt” – 312.35 (300)
“600h – 300 gener R1b 111 mrk.txt” – 310.61 (300)