It is about 20 months as NevGen YDNA Haplogroup Predictor started his life on internet, on www.nevgen.org. During that time, several people, mostly from universities across the world, asked us for desktop version. Also, some of them asked for batch capability, for quick processing of hundreds of haplotypes from academic works of different world populations. So, we decided to come in their way and release free desktop edition of predictor, with capability for Promega and YFiler sets of 23 or 17 markers, since most academic haplotype collections are done with that marker sets, together with batch processing capability.
First edition contains only haplogroup predictor, but soon it should contain several other tools, some for Y-DNA, some for autosomal data (that is why it is called “NevGen Genealogy Tools”). This tools have been written several years ago, some of them before predictor, but time is needed to prepare them for english-speaking world, with better translations and user manual. For now desktop program works in english language, soon we expect to be russian language too, as soon translation is finished.
In future, another levels might be added to predictor’s desktop edition, for now is only one available. Into available limited level for 23 markers could be entered other markers, but they shall be ignored.
Update of October 02, 2017. Now is available R1a Level in Predictor. Russian translation of program is here too. And, most important, “NevGen probability Calculator of Time to Most Recent Common Ancestor” is available to Genetic Genealogy community.
Update of January 15th, 2018. Now are available two newly added tools. First is “Viewer for Autosomal PCA charts”, with calculated PCA Chart of Europe, Near East and North Africa (first four axis). Second is Autosomal Admixture Calculator.
Also is now available E Level in Predictor.
Update of December 27th, 2018. Now is available J Level in Predictor.
There is no setup code for this, user only has to unpack all contents into the same folder, and to rename “NevGen.e” into “NevGen.exe”. Program is then simply started by executing “NevGen.exe”. Please keep in mind that any .dat files should be kept in the same directory as NevGen.exe. Otherwise, predictor shall not work.
Here is link to download NevGen Genealogy Tools v1.1.
This edition was helped by Serbian DNA Project and Radimpex Civil Engineering Software.
About batch capability and format of it’s input data
For 23 marker STR set (Promega), data should be entered in format like this (for this input, please select “23 markers Order (Promega)” in listbox at bottom of the dialog box):
; This row is comment, because it starts with semicolon sign (";"), and is meaningful only for batch input.
16 13 20 30 13 10 23 11 11 11 14 17 23 24 13 14 10 13 15 14 17 15 12 ; BO73 Bolivia [Native American]
19 13 19 29 14 11 22 13 12 12 15 18 23 24 12 13 10 13 16 11 13 16 12 ; ID Oviedo 269 Asturias, Spain [Spanish]
16 12 22 29 15 10 22 13 12 10 15 19 21 21 11 11 12 15 15 13 16 15 11 ; 62 Basel, Switzerland [Swiss]
Please note that in the same line with haplotype, after optional semicolon sign “;” any comment can be placed, which does not affect calculation, but might be very useful to user who is batch proccessing haplotype set. Without such possibility, work with it could be much harder. Every input row, regardless whether it contains haplotype or it is only comment, is copied into output. This is output of this batch proccesing:
; This row is comment, because it starts with semicolon sign (";"), and is meaningful only for batch input.
1. ----------
16 13 20 30 13 10 23 11 11 11 14 17 23 24 13 14 10 13 15 14 17 15 12 ; BO73 Bolivia [Native American]
Probability = 98.80% Fitness=37.34 [1.34] Q M346>> M3> M848
Probability = 1.19% Fitness=30.83 [0.93] Q M346>> Z780
2. ----------
19 13 19 29 14 11 22 13 12 12 15 18 23 24 12 13 10 13 16 11 13 16 12 ; ID Oviedo 269 Asturias, Spain [Spanish]
Probability = 100.00% Fitness=53.30 [1.31] R1b (for 67+ markers, try level for R1b-s, 300+ subclades)
3. ----------
16 12 22 29 15 10 22 13 12 10 15 19 21 21 11 11 12 15 15 13 16 15 11 ; 62 Basel, Switzerland [Swiss
Probability of unsupported subclade: 93.61%
Warning: Values of fitness (or relative fitness) are rather small, so results are not too confident. It is possible that its haplogroup is not supported by current version of predictor (so called "FALSE POSITIVE"), or haplotype really belongs to some supported haplogroup, but it is rare or too distant branch, which is not sufficiently represented in samples used by predictor.
Probability = 5.87% Fitness=19.63 [0.48] G2a2b1 M406> FGC5081> Z17887
Probability = 0.38% Fitness=18.54 [0.44] G2a2b1 M406> PF3293
Probability = 0.12% Fitness=18.96 [0.39] G2a2b1 M406> FGC5081> L14
Probability = 0.03% Fitness=16.64 [0.40] G2a2b1 M406> M3302
Probability = 0.00% Fitness=14.64 [0.29] G2a2 > L497>> Z725>> L43
Probability = 0.00% Fitness=13.44 [0.44] G2a2a PF3147
Probability = 0.00% Fitness=12.37 [0.38] G2a1-L293
Probability = 0.00% Fitness=11.72 [0.37] G1 M342
Probability = 0.00% Fitness=11.79 [0.25] G2a2 > L497>> Z725>> CTS4803> S2808
Probability = 0.00% Fitness=11.71 [0.26] G2a2 > L497>> Z725>> CTS4803
Probability = 0.00% Fitness=11.26 [0.29] G2a2b2a1c - Z724
Probability = 0.00% Fitness=11.70 [0.30] I2a2b-L38
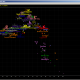
Output of batch prediction is automaticaly saved in file “Batch prediction.txt”, in folder where executable is positioned, in Unicode TXT format. This is how it looks in dialog box of predictor (drawn picture is of last haplotype in batch):

Please be aware that in picture in right side of the dialog box entered STRs are allways displayed in FTDNA order and FTDNA format for GATA H4, regardless of format of entered haplotype. Description of meaning of picture, and many other things (like SNP restriction), can be found here.
Button “Most frequent values” replaces entered haplotype with most frequent values for the first subclade selected in listbox in right upper corner, and redraws it in picture field. It does not display modal values (probable STR values of most recent common ancestor of subclade) for selected subclade, but the most frequent ones. It is not allways the same. It can be useful sometimes, mostly for comparing STR value statistics of different haplogroups (subbranches).
Should be kept in mind that this version ignores any incomplete values of STRs (for example 20.2 is treated like 20). If value of STR is unknown, use $ sign. For example:
16 13 20 30 $ 10 23 11 11 11 14 17 23 24 $ 14 10 13 15 14 17 15 12 ; BO73 Bolivia [Native American].
FTDNA order is the most general format of entering STR data into predictor, because it allows any STR marker to have more than one value, multiple values of the same STR are separated by ‘-‘ sign. In order of 23 or 17 markers (Promega and YFiler) having multiple values for any STR other than DYS385 can make problems, because they expect exact 23 (or 17) values in array. For example, having multiple marker value for DYS19 (it is common in some subclades of haplogroup G) would yield longer array, which would not be recognised by predictor. In such cases, when there are markers with multiple values other than DYS385 it is recommended to convert data from Promega and YFiler formats of 23 or 17 markers into more general FTDNA format.
Here is such sample, with values of 15 and 16 on DYS19.
19 14 22 30 15,16 10 21 12 10 10 16 18 24 21 11 11 10 14 18 13 14 15 14 ; E13-265 Basel, Switzerland [Swiss]
Reordering of STR values together with inserting of $ signs for unknown marker values can be easily done in any spreadsheet program. Beware that when converting STR data from Promega and YFiler formats of 23 or 17 markers into FTDNA format value od GATA H4 should be decreased by 1. When sample is reordered in that way, we got:
14 21 15-16 10 13-14 $ $ 11 14 11 30 18 $ $ $ $ 16 22 $ $ $ 13 $ 15 $ 19 18 $ $ 10 $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ 21 $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ 12 $ $ $ 10 $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ $ 24 $ 10 ; E13-265 Basel, Switzerland [Swiss]
Autosomal Admixture Calculator
This is just another utility which calculates percentages of admixture from autosomal genome data from FamilyTreeDNA Family Finder (.csv files) or 23AndMe (.txt files), using EM algorythm and using Calculator input files from Dodecad Ancestry Project (dodecad.blogspot.com). It is made in April 2014. There is not need for any conversion of format, you can just select your genotype file, ‘.par’ file of Calculator and press OK to calculate percentages.
Because we are not sure to have right to distribute Dodecad’s Calculator input files (they are property of Dienekes Pontikos), we de not include them in our Tools, but we give you links to pages where you can download them from dodecad.blogspot.com.
K7b and K12b calculators
http://dodecad.blogspot.rs/2012/01/k12b-and-k7b-calculators.html
There you can, in section “Downloads”, find links to get input files for both calculators.
globe13 calculator
http://dodecad.blogspot.rs/2012/10/globe13-calculator.html
world9 calculator
http://dodecad.blogspot.rs/2011/12/world9-calculator.html
globe10 calculator
http://dodecad.blogspot.rs/2012/10/globe10-calculator.html
weac2 calculator
http://dodecad.blogspot.rs/2012/06/weac2-calculator.html
K12a calculator
http://dienekes.blogspot.rs/2011/12/first-analysis-of-metspalu-et-al-2011.html
K10a calculator
http://dodecad.blogspot.rs/2012/06/k10a-calculator.html
africa9 calculator
http://dodecad.blogspot.rs/2011/09/africa9-calculator.html
dv3 calculator
http://dodecad.blogspot.rs/2011/09/do-it-yourself-dodecad-v-21.html
euro7 calculator
http://dodecad.blogspot.rs/2011/09/euro7-calculator.html
eurasia7 calculator
http://dodecad.blogspot.rs/2011/10/eurasia7-calculator.html
I believe previous list is not complete and there are some other useful Calculators which could be found on Dodecad Ancestry Project.
Calculation can also be restricted to any chromosome (by entering number of it), or part of it (by entering starting and ending base pair). While program calculates you could see displayed current results, and currently reached limit of calculation (termination condition, or Epsilon).
Here you can see dialog for entering input data for admixture calculation.

Here you can see dialog for displaying results of admixture calculation. In both dialogs is used K7b Calculator and ancient genome of Loschbour from Luxembourg.

We must warn users to keep all four Calculator input files (for example, ‘k12b.alleles’, ‘k12b.par’, ‘k12b.txt’ and ‘k12b.12.F’) in the same directory. Otherwise, it will not work.






Can request an addition to an R1a>Z93>FGC82884(and lower subclades) it is integral and most are now unified instead of being scattered under R1a>Z93
Hi,
As administrator of several I2a-L38 project I am an ethousiastic user of the Nevgen-predictor. To my pleasure I noticed that the Nevgen-predictor was used in a doctoral dissertation to determine the I-L38 haplogroup the aDNA Lichtenstein samples: https://ediss.uni-goettingen.de/handle/11858/13902?fbclid=IwAR1JSS5vQspStoPuwy9ECrh7ViCPAalq-YQ0LeS5Dgs818KVOWKAPg5gf4M
To convince the scientists to finetune this prediction to I2a-S2591 it would be great if I-L38 could be selected as a preselection.
My question: is it be possible to add I2a-L38 as an option in the settings-list (below I2a2…) and/or in the known SNP list of the nevgen-predictor?
Ps: If I can help with delivering data to finetune the I2a-L38 subclades, please let me know.
Best wishes,
Hans De Beule / hans.debeule@hotmail.com